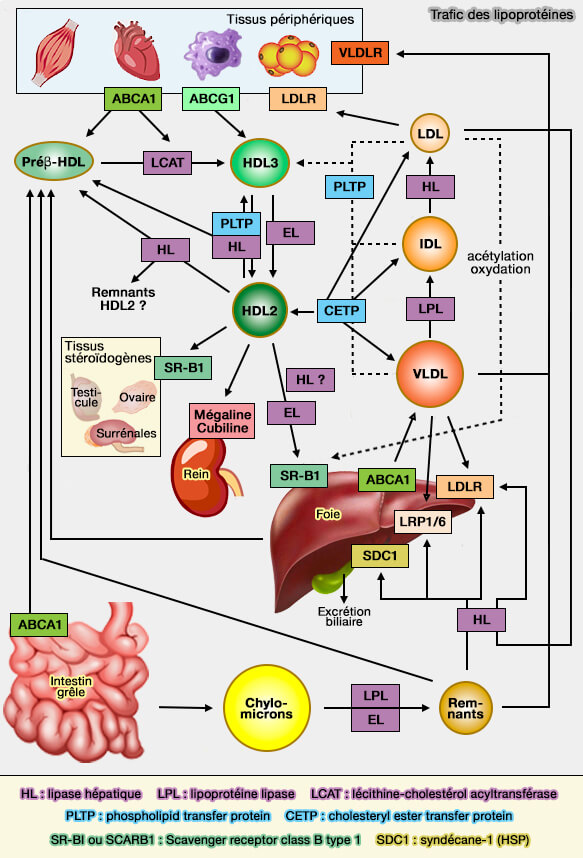
L'inactivation de l'expression de SDC1 est associée à des taux élevés de triglycérides plasmatiques en raison d'une altération de l'absorption hépatique résiduelle.
La régulation réciproque et coordonnée de LDLR et LRP1 avec SDC1 ne se produit apparemment pas, et contrairement à l'expression de LDLR et LRP1, l'expression de SDC1 n'est pas affectée par des altérations des taux de cholestérol circulant.
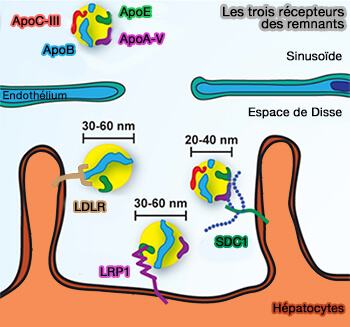
Récepteurs des remnants
(Figure : vetopsy.fr d'après Gordts et coll)
La livraison de lipoprotéines aux lysosomes est lente, i.e. demi-vie de 30–45 min par rapport aux récepteurs à internalisation rapide, i.e. LDLR, 10 min et LRP1 30 sec.
Toutefois, la capacité de liaison de SDC1 pour les remnants est 10 fois supérieure à celle de LDLR et LRP1 d'au moins un ordre de grandeur, probablement en raison de la multivalence offerte par les multiples chaînes d'héparane sulfate (HS) sur SDC1.
Dans des conditions de jeûne, l'occupation de SDC1 est probablement inférieure à 10% de sa capacité, alors que LDLR et LRP1 peuvent être saturés via la liaison TRL et LDL.
L'élimination de l'apoA-V des TRL inhibe la liaison au HS de SDC1 et bloque l'absorption des TRL.
L'absence d'apoE ou d'apoA-V entraîne une accumulation de petits TRL dans la circulation, ce qui est corrélé au développement accru de l'athérosclérose.
b. L'apoC-III s'accumule sur les TRL plasmatiques chez les souris dépourvues de SDC1 hépatique, mais pas chez les souris dépourvues de LDLR et/ou de LRP1.
SDC1 est le principal récepteur responsable de la clairance des TRL riches en apoC-III, ce qui concorde avec l'observation selon laquelle les souris exprimant SDC1 fonctionnel ont très peu d'apoC-III sur les TRL circulants.
Remarque : le DT2 augmente également l'expression et la sécrétion de SULF2 dans les hépatocytes, réduisant la 6-O-sulfatation des HS. La combinaison de ces facteurs avec une production accrue de VLDL (lipoprotéines de très basse densité) et une translocation altérée de LRP1 pourrait expliquer l'hypertriglycéridémie sévère souvent manifestée par les patients atteints de DT2.
SDC1 est responsable de l'élimination des petits TRL, i.e. les plus athérogènes.
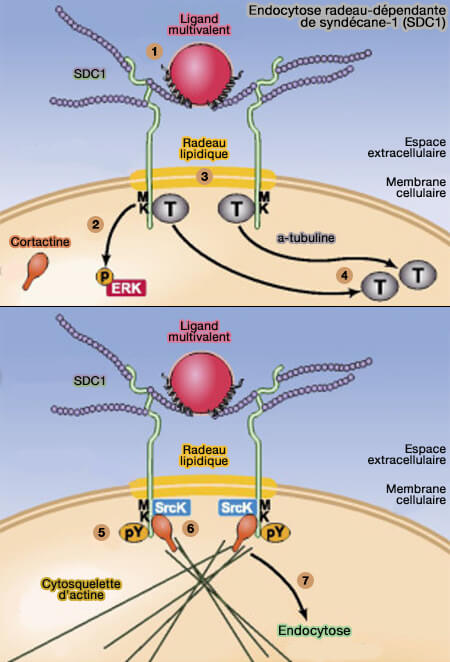
Endocytose radeau-dépendante de syndécane-1 (SDC1)
(Figure : vetopsy.fr d'après Chen et coll)
a. La phase initiale rapide de l'endocytose SDC1 est déclenchée par la liaison du ligand (1) qui déclenche une activation rapide de ERK (2) et le regroupement entraîne le mouvement du syndécane-1 dans les radeaux (3).
Le radeau lipidique nécessite des microdomaines membranaires riches en cholestérol indépendamment des autres molécules intervenant dans le mécanisme.
L'activation de ERK entraîne la dissociation du syndécane-1 de l'α-tubuline (T), une molécule qui, pourrait servir d'ancrage pour le syndécane-1 au niveau de la membrane plasmique à l'état basal (4).
b. La seconde phase de l'endocytose de SDC1 est provoquée par le regroupement du motif MKKK cytoplasmique qui déclenche progressivement la phosphorylation dépendante de la kinase Src des résidus tyrosine conservés dans les domaines transmembranaire et cytoplasmique de SDC1 (5).
Cette phosphorylation recrute la cortactine (6), puis l'endocytose efficace actine-dépendante (7).
Remarque : le motif MKKK est également présent dans SDC3 et SDC4 (mais pas SDC2), ce qui suggère que d'autres éléments régulateurs, ou sa localisation et/ou son niveau d'expression différencient SDC1 en tant que récepteur de clairance HSPG dominant.
Autres HSPG impliqués dans le trafic des lipopprotéines
les HSPG dans le piégeage ou la rétention des lipoprotéines contenant des apoB riches en cholestérol, i.e. ce modèle est appelé " réponse à la rétention " dans les lésions vasculaires.
L'athérosclérose est étudiée succinctement dans un chapitre spécifique.





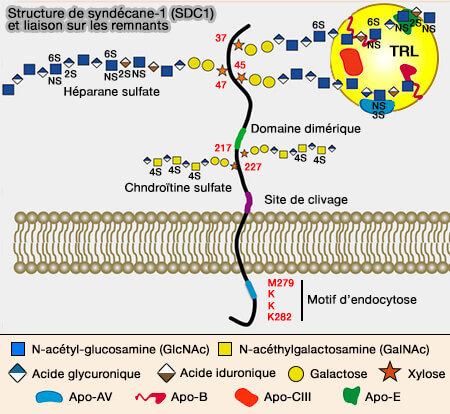
 structure générale des syndécanes).
structure générale des syndécanes).